|






 |  |

| Medicina | Medicina veterinara | Muzica | Psihologie | Retete | Sport |
Medicina
|
|
Qdidactic » sanatate & sport » medicina Boli genetice si anomalii congenitale cu depistare neonatala |
Boli genetice si anomalii congenitale cu depistare neonatala
Boli genetice si anomalii congenitale cu depistare neonatala
Anomalii genetice
Anomaliile monogenice, cauzate de o singura gena mutanta se intalnesc la 0,20% de nascuti vii si constituie in jur de 7,5% din totalul malformatiilor manifeste la nastere.
Mutatia genica este o alterare a structurii chimice a moleculei de ADN prin aditia, deletia sau inversia unui nucleotid si este urmarea agresiunii unor factori din mediul extern, (ex.radiatiile ionizante, factorul mutagen cel mai bine studiat). In raport cu celula care a suferit mutatia, se poate distinge mutatia somatica si gametica.
Mutatia somatica vizeaza afectarea materialului genetic dintr-o celula obisnuita a corpului, mutatie care se transmite la celulele fiice si formeaza o linie celulara noua (mozaicism). In aceste cazuri, boala nu se transmite la descendenti, cu moartea bolnavului mutatiile somatice isi pierd importanta biologica si sociala.
Mutatia gametica apare cand agresiunea s-a produs asupra celulelor de reproducere. Persoana care a suferit mutatia gametica nu manifesta nici un simptom de boala, dar mutatia se transmite la urmasi. Cand mutatia se gaseste la un singur gamet numai un urmas va fi malformat, iar anomalia intereseaza toate celulele corpului, inclusiv cele de reproducere. Pericolul acestei mutatii consta in posibilitatea raspandirii ei la restul populatiei prin mostenire (ereditar). Boala familiala este maladia care se observa la mai multi membri ai unei familii si poate fi ereditara sau castigata prin: infectii, toxice, carente nutritionale, izoimunizare etc. Fiecare gena ocupa in cromozom un locus si are o gena analoga in cromozomul pereche. Cele doua locusuri pereche se numesc omologe iar genele se numesc alele. Genele alelice sunt analoge dar nu sunt identice.
Cand individul are o gena mutanta si o gena alela normala se numeste heterozigot, iar cand ambele gene sunt mutante homozigot. Transmiterea la descendenti a unei anomalii cauzata de o singura gena mutanta se face dupa legile mendeliene. Dupa aceste legi, o gena mutanta se poate transmite autozomal-dominant, autozomal-recesiv, dominant -X- linkata sau recesiv -X- linkata.
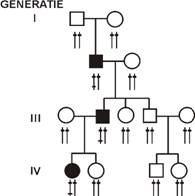
Transmiterea este autozomal-dominanta cand o singura gena mutanta, localizata la nivelul cromozomilor autozomali poate determina manifestarea clinica a anomaliei, adica gena mutanta se manifesta pe plan clinic in starea heterozigota. Se cunosc in jur de 943 de boli ereditare cu transmitere dominanta si cu gena localizata la nivelul cromozomilor autozomali. Caracteristic pentru anomalia cu transmitere dominanta este faptul ca pe arborele genealogic este asezata vertical si fara intrerupere (Figura 1). Bolnavul transmite boala la 50% dintre copii.
|
LEGENDA ↑↑ o pereche de autozomi ↨ o gena anormala □ Barbat ○ Femeie ■ ● Individ afectat |
|
Fig.1 Transmitere autozomal-dominanta. |
Aceasta continuitate ascendent proband descendent poate lipsi cand este vorba de o anomalie indusa sporadic, printr-o mutatie noua .
Intre bolile ereditare, aproximativ 783 se transmit recesiv, ceea ce inseamna ca pentru a apare manifestarea clinica de boala este nevoie ca gena mutanta sa fie in doza dubla, adica ambele gene ale unei perechi de cromozomi sa fie mutante (stare homozigota). Cand genele unei perechi de cromozomi difera, una este mutanta si cealalta normala (stare heterozigota) boala nu se manifesta, ramane ascunsa din punct de vedere clinic (recesiva).
In general, majoritatea bolilor autozomal-recesive (Figura 2) sunt fara istoric familial deoarece, de obicei parintii bolnavi sunt heterozigoti, adica fenotipic sanatosi. Descendentii parintilor heterozigoti vor fi: afectati in proportie de 25%; neafectati, dar purtatori de gena mutanta 50% si sanatosi 25%. Realizarea acestei probabilitati este mica din cauza numarului mic de copii dintr-o familie.
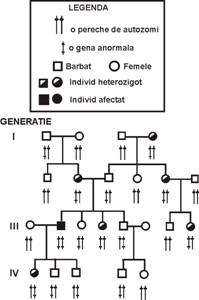
Fig.2 Transmitere autozomal-recesiva.
O alta caracteristica a bolilor autozomal-recesive consta in aceea ca urmasul probandului (bolnavului), in general este sanatos, deoarece aproape intotdeauna acesta se casatoreste cu persoane indemne. Frecventa bolilor recesive creste in cazul casatoriilor consanguine si la grupurile de populatie izolata.
Intre bolile ereditare, nu toate genele mutante se gasesc pe cromozomii autosomali; se descriu in jur de 150 de anomalii congenitale cu mostenire legata de cromozomul X (X-linkata). Cunoasterea malformatiilor cu mostenire legata de sex, este importanta deoarece stabilirea prenatala a sexului ne poate permite descifrarea unor atitudini de profilaxie prenatala. O particularitate a bolilor cu transmitere recesiva legata de sex (Figura 3 si 4) este ca acestea se manifesta mai frecvent la baieti decat la fetite. Asa este cazul hemofiliei. Explicatia frecventei scazute la fetite consta in probabilitatea mai mica de a se intalni gena mutanta pe ambii cromozomi sexuali, XX. Explicatia frecventei crescute in cazul individului de sex masculin consta in aceea ca cromozomul Y are numai gene de sexualizare si ii lipseste astfel gena pereche sanatoasa cu rol in neutralizarea genei patogene de pe cromozomul X. Procesele actuale ale geneticii fac ca numarul de anomalii genetice descoperite sa fie in crestere sporind astfel ponderea geneticii clinice in patologia umana.
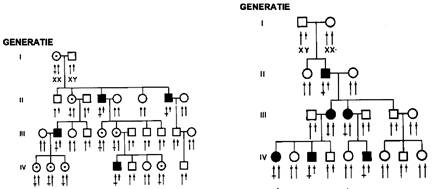
|
Fig. 3-4: 3. Transmitere recesiva X-linkata ; 4.Transmitere dominanta X-linkata. |
Anomaliile cromozomiale ca si cele genice, pot fi consecinta unei transmiteri ereditare, dar sunt mai ales consecinta unor mutatii noi, mai extinse, ce constau din nesepararea unui cromozom cu pierderea fragmentului sau reanexarea lui la acelasi cromozom sau la altul (Figura 5): Atunci cand nesepararea cromozomilor survine in timpul diviziunii mitotice a celulelor embrionare, viitorul copil va avea conformatia genetica anormala numai in anumite zone ale organismului (mozaic). Aceasta forma de anomalie, mozaicism, este mult mai rar intalnita.
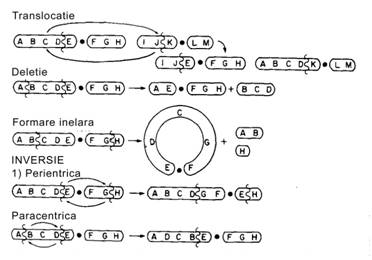
Fig.5 Mecanisme de formare a aberatiilor cromozomiale.
Anomaliile cromozomiale se intalnesc la 0,60 % de nou-nascuti vii, dar numai o parte dintre acestia prezinta anomalii fizice manifeste la nastere. Prezentam in continuare succint, in ordinea frecventei principalele anomalii cromozomiale insotite de anomalii fizice, manifetate sau depistate la nastere. Prezenta unei anomalii fizice sau a unei asocieri de anomalii fizice la nastere (sub genericul "sindrom dismorfic") trebuie sa conduca neonatologul la solicitarea unui examen genetic si a unui cariotip.
Trisomia 21 (Sindromul Down, mongoloismul)
Incidenta sindromului Down este de aproximativ un caz la 600-700 de nou-nascuti vii. Substratul citogenetic al bolii este o aberatie numerica sau structurala autozomala cu surplus de cromatina. Fenotipul sau manifestarea clinica a bolii consta dintr-o dismoprfie caracteristica a fetei si corpului la care, in general se asociaza o retardare mintala.
Cele mai numeroase cazuri de sindrom Down se caracterizeaza din punct de vedere citogenetic,prin prezenta unui cromozom supranumerar la perechea 21, iar cariotipul acestora se exprima prin formula 47 XY sau 47XX.
Aceasta forma de anomalie numerica autozomala apare sporadic si este strans legata de varsta mai avansata a gravidelor, ceea ce pledeaza pentru un determinism legat indeosebi de factorii de mediu extern. Afectiunea se poate transmite urmasilor dar din fericire, foarte putini bolnavi se casatoresc si ajung sa dea nastere la copii. Patogenia trisomiei 21 are la baza nondisjunctia perechii a 21-a de cromozomi in timpul meiozei. Perturbarea separarii acestei perechi pare a fi in legatura cu actiunea nociva a razelor X sau a unor virusuri. Nesepararea da nastere unui ovul sau spermatozoid cu doi cromozomi 21, care dupa fertilizare cu un spermatozoid sau ovul normal, va da nastere unui zigot cu trei cromozomi 21 (trisomie). O alta forma de trisomie 21 cu fenotip de sindrom Down uneori complet alteori incomplet sau chiar normal este mozaicismul. Aceste anomalii se datoreaza nondisjunctiei sau aberatiilor de structura survenite dupa primele diviziuni ale oului. Aceasta face ca numai o parte din celulele organismului sa fie afectate. In cazurile in care nu este afectat ectodermul din care ia nastere sistemul nervos, dezvoltarea intelectuala ulterioara se desfasoara normal. Cariotipul cazurilor de mozaicism este 47XX+21/46XX .
Simptome. In general, defectele anatomice permit de la prima observatie clinica suspectarea sindromului Down. Dintre acestea vom aminti cateva in cele ce urmeaza, cu mentiunea ca nu este obligatorie prezenta tuturor la fiecare caz in parte:
Greutatea la nastere, in general, este mica.
Craniul este mai mic, prezinta brahicefalie si un perimetru suboccipito-bregmatic mai mic. Gatul este mai scurt si cu piele in exces la nivelul cefei.
Ochii sunt mai departati unul de celalalt (hipertelorism), iar baza nasului este aplatizata, din cauza hipoplaziei piramidelor nazale. Fanta palpebrala este oblica de sus in jos si in afara. In unghiul intern al ochiului se poate observa a 3-a pleoapa (epicantus), un pliu pe 1/2 interioara a pleoapei superioare.
Gura este intredeschisa, iar limba propulseaza in afara cavitatii bucale. Miscarile limbii si ale buzelor realizeaza o grimasa destul de caracteristica. Palatul este ingust si scurt.
Musculatura este hipotona. Hipotonia musculara si hiperlaxitatea ligamentara permit efectuarea unor miscari pasive foarte ample ale extremitatilor.
Mainile
au un aspect caracteristic: degetele sunt mai scurte si relativ egale,
ceea ce da impresia de mana patrata. Una din falangele
degetului mic poate lipsi dand numai un sant de flexiune pe fata
palmara a degetului.
La nivelul piciorului se poate remarca intre degetul mare si urmatorul un spatiu marit si un sant adanc care de aici incepe si merge spre marginea interna a plantei facand un arc in jurul eminentei tenare.
Retardare in dezvoltarea psiho-motrica.
Se mai pot asocia: cardiopatie congenitala (comunicare interventriculara), atrezie duodenala si uneori leucemie.
Prognosticul este, in general, nefavorabil, decesul survine la aproximativ 90% din cazuri, pana la varsta de 20 de ani.
Trisomia 18.
Trisomia 18 are patogenie asemanatoare trisomiei 21. Afecteaza mai frecvent fetitele. Varsta parintilor este mai inaintata. De regula, decesul survine in primele luni dupa nastere prin insuficienta cardiaca pe fond de cardiopatie congenitala.
Trisomia 18 se manifesta prin urmatoarele simptome: fatul are greutatea la nastere mai mica decat varsta gestationala, craniul mic, occiputul proeminent, micrognatie, microstomie, microftalmie, comunicare interventriculara sau canal arterial, degete permanent flectate si puternic distantat aratatorul de medius, dermatoglife anormale, (de regula arcuri), plica simiana, picior scobit sau varus equin, atrezie biliara si retardare mintala. In afara trisomiei 18, malformatii asemanatoare genereaza deletia bratului lung sau scurt al cromozomului 18.
|
Trisomia 13.
Trisomia 13 sau boala lui Patau se intalneste foarte rar si decesul survine pana la varsta de 1 an. Bolnavul prezinta: microcefalie, fruntea tesita, microftalmie sau anoftalmie, urechi jos inserate, intarziere mintala, convulsii, malformatii cardiace, criptorhidie, anomalii ale degetelor.
Deletia bratului scurt al cromozomului 5.
Simptomele acestei aberatii structurale a cromozomului 5 sunt: microcefalia, micrognatia, fanta palpebrala oblica, hipertelorism, epicantus, retardare mintala, malformatie cardiaca. Caracteristic este strigatul asemanator mieunatului de pisica ("cri du chat").
Sindromul Turner
Sindromul Turner sau disgenezia gonadala are o frecventa de 1 la 2-3 mii de fetite nou-nascute si 1 la 20 de avorturi. La baza disgeneziei gonadale si a anomaliilor dismorfice care caracterizeaza pe plan clinic sindromul Turner sta lipsa unui cromozom sexual din cariotip sau aberatia structurala a cromozomului X.
Sindromul Turner poate fi cauzat de absenta unui cromozom X din cariotipul feminin, de un defect de structura a acestuia sau de absenta cromozomului Y din cariotipul masculin. Aberatiile numerice se stie ca apar in timpul formarii celulelor primare sexuale sau in timpul diviziunii mitotice a zigotului. In primul caz, cariotipul este 45X iar in al doilea caz poate fi 45X/46XX, 45X/47XXX (mozaicism). Mozaicismul, care include si cromozomul Y, se intalneste foarte rar si poate avea cariotipul 45X/46XY sau 45X/47XYY. Dintre numeroasele aberatii numerice posibile, forma cu cariotipul 45X se intalneste la peste 85% din totalul cazurilor de sindrom Turner. Cele mai obisnuite anomalii structurale generatoare de sindrom Turner sunt izocromozomul si deletia bratului lung sau scurt al cromozomului X.
Anomaliile gonozomale mentionate cauzeaza malformatii congenitale si anomalii ale cresterii. Intre malformatii, in mod constant, se semnaleaza absenta gonadelor sau gonade rudimentare, ceea ce atesta si denumirea de disgenezie gonadala.
Sindromul Turner poate fi diagnosticat clinic de la nastere sau mai tarziu pana la varsta pubertatii.
Forma cu manifestare precoce se mai numeste sindrom Bonnevie-Ullrich si consta din: limfedem la nivelul dosului picioarelor si mainilor, gat palmat (pterigium coli), talie mica pentru varsta gestationala si coarctatie de aorta la un nou-nascut cu fenotip de regula feminin. Mai pot fi prezente si alte anomalii ca: micrognatie, cubitus valgus, urechi jos implantate, cataracta, epicantus, hipertelorism, cutis laxa etc.
Daca simptomele sindromului Bonnevie-Ulrich nu se observa la nastere, diagnosticul se stabileste mai tarziu, cand parintii aduc la medic copilul pentru intarziere accentuata a cresterii. Diagnosticul este mai sugestiv cand aceasta crestere intarziata survine la un copil cu unele din semnele dismorfice mentionate.
Odata cu inaintarea in varsta, spre pubertate, diagnosticul devine mai evident datorita manifestarilor disgeneziei ovariene: infantilismul organelor genitale, amenoree primara, mamele hipoplazice, mult departate una de alta, pilozitate redusa si de tip viriloid si infertilitate. La acestea se pot asocia si alte anomalii ca surditatea si retardarea mintala.
Majoritatea pacientilor cu sindrom Turner au modelul de cariotip 45X/46XX (mozaicism). Aceasta face ca simptomele clinice sa fie mai atenuate semnalandu-se aparitia caracterelor sexuale secundare si chiar fertilitate.
Sindromul Turner la persoanele de sex masculin (45X/46XY) se intalneste foarte rar, se asociaza de regula cu criptorhidrie sau alte forme de hipomasculinizare in raport cu gradul aplaziei testiculare. Cand aplazia este completa organele genitale sunt feminine. Statura mica si alte anomalii somatice pot lipsi. De mentionat aici este faptul ca gonadele vestigiale au tendinta la malignizare, ceea ce impune interventia operatorie pentru inlaturarea lor.
Diagnosticul de sindrom Turner se confirma prin demonstrarea negativitatii cromatinei sexuale din frotiul bucal si prezenta unui singur cromozom X in cariotip. Mozaicul si anomaliile de structura se confirma numai prin efectuarea cariotipului. Secretia hormonului foliculino-stimulator (HFS) este normala in perioada neonatala si crescuta ulterior, ceea ce permite deosebirea de nanismul hipofizar in care secretia este scazuta.
Sindromul Klinefelter
Sindromul Klinefelter este cauzat de un surplus de gonosomi X si Y la persoane cu fenotip masculin. Este una din cele mai frecvente anomalii cromozomiale, avand o incidenta de 1 la 500 de nou-nascuti de sex masculin. Intre avortoni, incidenta anomaliei este rara. Cea mai frecventa formula cromozomiala este 47XXY si mai rar 48XXYY, 48XXXY, 49XXXXY. Una dintre cauzele favorizante ale nondisjunctiei meiotice sau mitotice este, ca in sindromul Down, varsta inaintata a mamei.
Simptomele apar in mod obisnuit la pubertate si constau din: talie mica, ginecomastie, testiculi mici, atrofiati, cu tubi seminiferi hialinizati, infertilitate prin azoospermie si retardare mintala. Incidenta retardarii mintale la cazurile de sindrom Klinefelter creste cu cresterea numarului de cromozomi X din cariotip, iar incidenta criminalilor cu cresterea numarului de cromozomi Y. Depistarea cazurilor de Klinefelter, in perioada neonatala, se poate face cu ajutorul testului Barr in cadrul screeningului de masa.
Anomalii peristazice
Cauze legate de mediu (factori de mediu teratogeni). Cu toate ca numarul factorilor teratogeni siguri sau probabili este in crestere, acestia nu cauzeaza decat 4,36% din totalul malformatiilor congenitale majore. Bolile materne preexistente sarcinii sau care survin in timpul sarcinii cauzeaza 3,5% din totalul malformatiilor congenitale.
Infectiile, (in special rubeola, toxoplasmoza si citomegalia), cauzeaza peste 2% din totalul malformatiilor congenitale majore. Hipertermia, indiferent de origine, poate cauza anomalii ale extremitatii cefalice (anencefalia, microcefalia, microftalmia), probabil prin intermediul unei carente acute de acid folic. Bolile endocrine si metabolice ale mamei cauzeaza: prin diabet 1,44% din ansamblul malformatiilor congenitale (9% din copiii mamelor diabetice prezinta malformatii), iar prin hipotiroidism, fenilcetonurie s.a. un procent nesemnificativ. Cauze mecanice: prezentatia pelviana poate induce displazia de sold si malformatia piciorului; oligohidramniosul, hipoplazie pulmonara si malformatia piciorului; bridele amniotice, amputatii, stricturi, despicaturi ale fetei, si gastroschizis, iar presiunea hidraulica crescuta favorizeaza aparitia artrogripozei prin interferarea intoarcerii venoase de la nivelul membrelor inferioare.
S-a dat denumirea de disruptie defectelor structurale rezultate din distrugerea unei structuri sau parti embrionare initial normal formate.
Cauzele disruptiei sunt: anomalia sacului amniotic, obstructii vasculare prin emboli sau trombi si anastomoze vasculare. Bridele si fisurile amniotice cauzeaza stricturi si amputatii ale unor parti embrionare. Cu cat ruperea sacului vitelin este mai precoce cu atat gravitatea anomaliilor este mai mare.
Ruperea sacului amniotic la 3 saptamani de gestatie poate cauza: anencefalia, despicatura fetei, encefalocelul, meningocelul; la 5 saptamani: atrezia coanala, despicatura buzei superioare, polidactilia, sindactilia, defecte ale peretelui abdominal si toracic, scolioza; la 7 saptamani: palatoschizis, craniostenoza, benzi amniotice, stricturi, amputatii, limfedem, luxatia de sold, deformarea piciorului, scurtarea cordonului ombilical, iar la mijlocul gestatiei deformari caracteristice oligohidramniosului. Aproximativ 4% din cazurile cu anencefalie sunt consecinta ruperii precoce a sacului amniotic.
Interconexiunile vasculare se gasesc la gemenii monocorionici (placenta unica). In cazul suntului arterio-arterial sangele de la donator ce patrunde in vasele iliace ale primitorului determina o mai buna perfuzie a partii inferioare a corpului decat a partii superioare. La partea mai putin irigata a corpului (extremitatea cefalica, membrele superioare, cord, plamani, pancreas, intestinul superior) apar anomalii prin disruptie si morfogeneza incompleta. Donatorul poate prezenta: cardiomegalie prin suprasolicitare, insuficienta cardiaca cu disfunctie hepatica, hipoalbuminemie, edeme si progresiune spre hidrops. In cazul suntului arterio-venos cu debit mare, primitorul va prezenta: hipervolemie, cardiomegalie, poliglobulie, macrosomie, nefromegalie cu poliurie si polihidramnios, iar donatorul va prezenta: hipovolemie, rinichi mici, flux renal scazut, oligoamnios (cand gemenii sunt biamniotici) si hipotrofie.
In cazul unui geaman mort trecerea de la acesta a tromboplastinei sau a unor emboli prin placenta spre geamanul viu poate cauza acestuia, prin obstructii vasculare: ischemie si necroza, amputatii ale membrelor, hidranencefalie, microcefalie, atrezie intestinala, microstomie hemifaciala, gastroschizis, aplazie cutanata. Malpozitia unor parti fetale in uter poate cauza deformari ale toracelui sau cutiei craniene.
Teratogeni chimici Tutunul creste riscul malformatiilor cand numarul tigarilor fumate este mai mare de 10 pe zi. Alcoolismul poate produce variate malformatii prin intermediul acetaldehidei, primul produs catabolic al alcoolului etilic, ce rezulta sub actiunea alcool-dehidrogenazei si care are actiune toxica asupra embrionului si fatului. Dintre drogurile considerate teratogene pentru fatul uman amintim in ordine descrescanda a certitudinii: thalidomida, citostaticele, anticoagu-lantele (warfarina, dicumarol), iodurile, propiltiouracilul, dietilstilbestrolul, anticonvulsivantele (fenobarbital, hidantoina, trimetadiona) si progesta-tivele. Iodurile si propiltiouracilul determina hipotiroidism, dietilstil-bestrolul si progestativele anomalii genitale, iar restul drogurilor produc anomalii multiple. Streptomicina si chinina pot cauza leziuni ale nervului optic si auditiv, vitamina D stenoza aortica supravalvulara, iar litiul poate cauza anomalii ale cordului.
Dintre poluantii mediului extern, teratogene sunt unele ierbicide (ac. 2,4-diclorofenoxi-acetic) si mercurul.
Radiatiile ionizante. Cea mai constanta malformatie pe care o produc razele X este microcefalia. Sarcina iradiata cu mai putin de 1 rad are risc teratogen neglijabil, iar cand este iradiata cu mai mult de 10r (100 miliray) riscul este mare. In cazul catorva radiografii, embrionul primeste 10 miliray, iar cand se adauga si o radioscopie, embrionul primeste 100 miliray.
Fiziopatologie. Efectul teratogenilor depinde in mare masura de genotipul embrionului si al mamei, stadiul gestatiei si doza. Efectul legat de genotip poate fi ilustrat de actiunea diferita a teratogenilor la specii si subspecii diferite. Specificitatea teratogena de specie si subspecie este cauzata de deosebirile in ceea ce priveste capacitatea de metabolizare sau neutralizare a agentilor teratogeni.
In raport cu varsta gestatiei se semnaleaza susceptibilitate teratogena maxima in perioada primelor 2 luni, cand activitatea metabolica si proliferativa celulara este mare, in legatura cu geneza schitelor embrionare ale organelor. Cele mai expuse schite embrionare sunt cele de origine neuroectodermica. Schitele de origine mezodermica si endodermica sunt mai refractare la procesul teratogen.
Astazi se cunoaste un orar teratogenetic legat de data cand actioneaza factorii patogeni. Dupa mecanismul si data cand actioneaza acesti factori asupra produsului de conceptie, Smith distinge mai multe tipuri de malformatii (Tabel I).
Perturbarea morfogenezei normale a produsului de conceptie poate distinge 4 ipostaze:
Morfogeneza incompleta. In acest cadru se includ malformatiile ce rezulta din dezvoltarea incompleta a unei structuri. Astfel poate apare agenezia unui organ, hipoplazia (micrognatie), separarea incompleta (sindactilie cutanata), inchiderea incompleta (fanta palatina).
Morfogeneza aberanta. In acest caz exista o structura care s-a format anormal.
Morfogeneza accesorie. Acest tip de anomalie cuprinde structuri normal formate dar in exces (degete supranumerare, polisplenie).
Hamartomele. Reprezinta o tulburare de organizare embriogenetica ce face sa apara un tesut normal unde n-ar trebui sa fie si se manifesta adesea sub forma unei tumori.
|
Varsta cand actioneaza factorii patogeni |
Felul malformatiilor |
|
Perioada embrionara |
|
|
23 zile |
Ciclopie, holoprozencefalie |
|
|
Sirenomelie |
|
26 zile |
Anencefalie |
|
28 zile |
Mielomeningocel |
|
30 zile |
Atrezie esofagiana cu fistula traheoesofagiana |
|
34 zile |
Transpozitia vaselor mari |
|
36 zile |
Fanta labiala |
|
8 saptamani |
Atrezie rectala |
|
|
Hernie diafragmatica |
|
|
Comunicare interventriculara |
|
|
Sindactilie |
|
7-8 saptamani |
Atrezie duodenala |
|
8 saptamani |
Sinus sau chist branhial |
|
10 saptamani |
Fanta palatina |
|
|
Malrotatia intestinului |
|
|
Omfalocel |
|
|
Diverticul Meckel |
|
|
Uter bicorn |
|
Perioada fetala |
|
|
12 saptamani |
Hipospadias |
|
7-9 luni |
Criptorhidie |
|
9-10 luni |
Persistenta canalului arterial |
Tab.I - Anomalii in raport cu data actiunii factorilor patogeni (dupa Smith).
Anomalii multifactoriale
Cercetari epidemiologice si familiale ca si evaluarea matematica a datelor genetice ale scolii engleze au gasit explicatii unui mare numar de malformatii congenitale prin conturarea modului multifactorial. Malformatiile multifactoriale reprezinta efectul cumulativ al mai multor gene anormale mostenite si al agresiunii unor factori din mediul exterior. Numarul genelor implicate, natura factorilor de mediu si modul de transmitere la descendenti nu se cunosc.
Malformatiile multifactoriale constituie cea mai mare parte din totalul malformatiilor congenitale. Incidenta lor este de 0,7 % de nou-nascuti vii si constituie 36% din malformatiile congenitale majore. Cele mai obisnuite exemple de malformatii multifactoriale sunt: anencefalia, spina bifida, luxatia congenitala de sold, stenoza pilorica, despicatura de buza si palat, cardiopatia congenitala, hernia diafragmatica, hidrocefalia congenitala, boala Hirschprung etc..
O sinteza a incidentei malformatiilor congenitale dupa cauza poate fi ilustrata de statistica de la Boston Hospital for Women pe anii 1972-1975 pe un total de 18.155 nou-nascuti cu varsta gestationala mai mare de 20 saptamani (Tabel II):
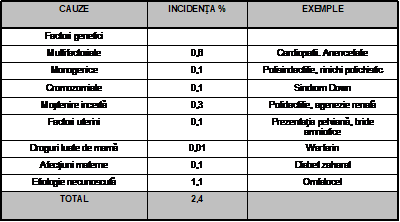
Tab.II. Incidenta malformatiilor congenitale dupa cauza ("Boston Hospital for Women").
CONSIDERATII CU PRIVIRE
LA ATIT
IN FATA UNEI MALFORMATII CONGENITALE. SFATUL GENETIC
In fata unui nou-nascut cu malformatii congenitale se vor lua urmatoarele masuri:
Se descriu in Foaia de Observatie amanuntit anomaliile, cu mentiuni exacte, pentru a se putea face comparatie cu valorile normale. Datele normale pentru comparatie vizeaza: talia, greutatea, raportul dintre diferite segmente ale corpului, perimetrul cranian, lungimea urechii, plasarea urechii, distanta intercantala, distanta interpupilara, distanta nazo-labiala, distanta inter-mamelonara, perimetrul toracic, lungimea mainii, a palmei, a degetelor, caracterul dermatoglifelor si o eventuala cataracta congenitala. Perimetrul cranian mic si hipotonia musculara pot fi revelatorii pentru o anomalie a creierului. Anomalia urechii se asociaza frecvent cu anomalia rinichilor, anomalia dermatoglifelor se asociaza cu anomalii cromozomiale, iar cataracta poate fi consecinta unei infectii congenitale.
Se face o anamneza amanuntita. Varsta mamei peste 35 de ani creste riscul pentru malformatii prin nondisjunctie cum sunt: trisomia 13, 18, 21 si sindroamele XXX, XXY. La 90-95% dintre pacientii cu trisomie 21 eroarea de meioza este de origine materna. Varsta inaintata a tatalui creste riscul mutagen pentru afectiunile autozomal-dominante, cum ar fi acondroplazia. Diabetul matern, infectiile TORCH, polihidramniosul si oligohidramniosul pot cauza variate malformatii. Polihidramniosul poate fi cauzat de anencefalie, tulburari ale SNC si atrezia esofagiana prin incapacitatea fatului de a inghiti lichidul amniotic. Oligohidramniosul poate cauza hipo-plazia pulmonara, anomalii pozitionale ale membrelor si sindrom Potter. Se va ancheta cand si cate droguri a luat mama in timpul sarcinii. Dovedirea ca un drog este teratogen este dificila deoarece majoritatea drogurilor exercita efecte nocive numai la o minoritate de feti. O statistica pentru un anumit drog pe 30-40 mii de subiecti a aratat cum ridica incidenta malformatiilor congenitale cu numai 1%. Mai mult de 2 avorturi in antecedentele mamei creste suspiciunea unei translocatii echilibrate la unul dintre parinti, ceea ce impune efectuarea de analize citogenetice.
Consanguinitatea pozitiva la parinti sau anomalii similare in arborele genealogic ne ajuta sa stabilim modul de transmitere a unei anomalii (autozomal-dominanta cand anomalia apare la fiecare generatie cu exceptia mutatiilor noi, autozomal-recesiva cand anomalia apare la urmasi si obisnuit lipseste la parinti, X-linkata cand anomalia apare mai frecvent la baieti). Consangvinitatea cauzeaza boala prin faptul ca favorizeaza aparitia genei patogene in doza dubla. Cunoscand modul de transmitere a unei boli putem afla riscul recurentei (25% in cazurile autozomal-recesive si 50% in cazurile autozomal-dominante).
Se fac examinari de laborator. In cazul malformatiilor multiple se face cariotipul. Daca malformatul este nascut mort sau decedeaza imediat dupa nastere, se face si un examen complet. Acesta include fotografia, radiografia anomaliilor scheletale aparente si semnalarea unor anomalii minore exterioare. In caz de deces, se face autopsia si se iau probe pentru cariotip (fragment steril din tegument, splina, timus sau gonade in thioglicolat ; sangele de la muribunzi sau nascuti morti nu este bun pentru cariotip din cauza leucocitelor foarte putin viabile).
La cazurile cu microcefalie, macrocefalie, cataracta, hepatomegalie, hipotrofie fetala se fac examinari serologice pentru toxoplasmoza, rubeola, citomegalie. Nu se fac examinari serologice la anomalii structurale cum sunt meningocelul, buza de iepure, polidactilia, agenezia de radius.
Parintii vor fi informati cu privire la cauza malformatiei propriului copil. Li se va spune daca va fi afectat si neuropsihic, ce reparatii chirurgicale sunt posibile si cand se pot face, daca urmatorii copii pot fi afectati de aceeasi maladie si daca este posibil diagnosticul prenatal.
Pentru diagnosticul prenatal se utilizeaza ultrasonografia, testarea serica materna, amniocenteza, testarea biochimica si testarea moleculara. Ultrasonografia fetala poate evidentia anomalii ale tractului digestiv si urinar, displazii grave ale scheletului, anomalii cardiace. Amniocenteza, efectuata la 16-17 saptamani de gestatie permite analiza ADN, analiza cromozomiala, analize biochimice, dozarea alfa-feto-proteinei si enzimelor intestinale.
|
MALFORMATIA |
RISCUL RECURENTEI |
|
Anomalii cromozomiale |
|
|
Trisomia 21 |
|
|
Translocatie, inversiune mozaicism |
Purtator tata |
|
Purtator mama |
|
|
Anomalii monogenice |
|
|
Boala polichistica renala (autozomal-recesiva) |
|
|
Sindromul Holt-Oram (autozomal- dominanta) presupunand ca un parinte este afectat |
|
|
Sindromul Telecantus-hipospadias, x-linkata, dominanta-mama este purtatoare |
50% din baieti sunt afectati |
|
50% din fetite sunt purtatoare |
|
|
Anomalii multifactoriale |
|
|
Spina bifida Anencefalie Meningomielocel Trunchi arterial comun Defect de sept ventricular Cheilo-palatoschizis Hipospadias Picior stramb (varus equin) Hidrocefalie Displazie de sold Boala Hirschsprung Fistula esofago-traheala Hernie diafragmatica Stenoza pilorica |
7% dintre baieti |
Tab. III - Riscul recurentei malformative in sarcina.
Alfa-fetoproteina, estriolul neconjugat si gonadotropina corionica sunt scazute in sarcinile cu sindrom Down, in timp ce in anencefalie si meningomielocel, alfa-fetoproteina este crescuta. Biopunctia vilozitara este o alternativa a amniocentezei.
Cea mai importanta problema pentru parinti este modul de a se transmite anomalia si riscul recurentei la urmatorii copii. Tabelul III arata riscul recurentei in sarcinile urmatoare pentru cele mai obisnuite malformatii congenitale.
| Contact |- ia legatura cu noi -| | |
| Adauga document |- pune-ti documente online -| | |
| Termeni & conditii de utilizare |- politica de cookies si de confidentialitate -| | |
| Copyright © |- 2026 - Toate drepturile rezervate -| |
|
|
|||
|
|||
|
|||
Analize pe aceeasi tema | |||
|
| |||
|
|||
|
|
|||