|






 |  |

| Aeronautica | Comunicatii | Drept | Informatica | Nutritie | Sociologie |
| Tehnica mecanica |
Tehnica mecanica
|
|
Qdidactic » stiinta & tehnica » tehnica mecanica Separarea cromatografica |
Separarea cromatografica
SEPARAREA CROMATOGRAFICA
Cromatografia reuneste un grup important de metode de separare diverse care permit separarea, izolarea, identificarea si dozarea simultana a componentilor din amestecuri complexe pe baza pe repartitiei diferite a componentelor intre o faza stationara si una mobila. Separarea compusilor prin cromatografie se datoreaza vitezei de migrare diferita a acestora de-a lungul fazei stationare, care este determinata de modul de distributie a substantelor de separat intre cele doua faze precum si de viteza de deplasare a fazei mobile.
Faza stationara poate fi un solid adsorbant (cromatografie de adsorbtie), un lichid depus pe suport solid (de repartitie), un schimbator de ioni (cu schimb ionic) sau un gel (cromatografie de excluziune sterica). Faza mobila poate fi gaz (Ar, He, N2) sau lichid (solvent sau amestecuri de solventi de inalta puritate, solutii tampon sau CO2 in conditii supercritice). La intrarea in coloana cromatografica, faza mobila poarta numele de eluent sau developant, iar la iesire se numeste efluent sau eluat.
Cea mai utilizata clasificare a metodelor cromatografice este cea care are la baza natura si starea de agregare a fazelor mobila si stationara. Din punct de vedere al mecanismelor de separare, metodele cromatografice se grupeaza dupa principalele interactii fizico-chimice care apar intre faza stationara si componentele amestecului, in timpul procesului de sorbtie-desorbtie.
In tabelul 1 este schematizata clasificarea metodelor cromatografice in functie de natura fazei mobile, a fazei stationare si a mecanismului separarii.
Tabelul 1. Clasificarea metodelor cromatografice
|
Nr. crt. |
Faza mobila |
Faza stationara |
Mecansimul separarii |
Abreviere |
|
|
Gaz |
Solid |
Adsorbtie |
CGS |
|
|
Lichid depus pe suport inert |
Repartitie |
CGL |
|
|
|
Lichid |
Solid: - adsorbant -schimbator de ioni - gel poros |
Adsorbtie |
CLS |
|
|
Schimb ionic |
CSA CSC |
||
|
|
Permeatie |
CPG |
||
|
|
Lichid |
Repartitie: -faza normala -faza inversa |
CLL |
Sediul procesului de separare cromatografica este coloana cromatografica, spatiul in care se depune faza stationara. In functie de forma coloanei in care este depusa faza stationara se disting: cromatografia pe coloana inchisa, cu umplutura (CC); cromatografia in tub deschis (de obicei o coloana de dimensiuni capilare pe al carei perete, un lichid sau un solid activ joaca rol de faza stationara); cromatografia plana: cromatografia pe hartie (CH) si cromatografia pe strat subtire (CSS); cromatografia pe filament.
Metodele cromatografice pot fi utilizate in scop analitic sau preparativ.
Pentru efectuarea separarilor si analizelor cromatografice se parcurg urmatoarele stadii: 1)pregatirea probelor (extractie, purificare etc.); 2) aducerea in contact a amestecului de separat cu faza stationara (spotarea); 3) separarea cantitativa (developarea) componentelor prin eluare cu faza mobila; 4) detectia (identificarea sau revelarea) componentelor prin metode fizico-chimice adecvate.
1.1. Cromatografia pe coloana
Este cel mai vechi procedeu cromatografic (Tvet, 1906). Piesa esentiala in aceasta metoda de separare o constituie coloana cromatografica, confectionata dintr-un tub de sticla, prevazut la partea inferioara cu un robinet, ales astfel incat raportul dintre diametrul coloanei si lungimea sa sa fie de 1:5 pana la 1:20. In coloana se gaseste faza stationara, care este un material adsorbant poros (de ex.: silicagel, alumina, carbune, carbonat de calciu, amidon, zaharoza, etc.). La partea superioara a coloanei se ataseaza o palnie de separare cu solvent pentru elutie.
Modul de lucru Pentru pregatirea coloanei, la capatul inferior al coloanei se pune un dop mic din vata, dupa care coloana se umple cu adsorbanti activati in prealabil. Pentru depunerea omogena a adsorbantului, fara bule de aer, se poate recurge la trei procedee: 1) adsorbantul se toarna in stare uscata in tubul gol; 2) coloana se umple 50% cu solvent inert si se depune apoi adsorbantul, mentinand constant nivelul eluentului prin scurgerea concomitenta pe la partea inferioara; 3) adsorbantul se toarna sub forma unei suspensii fine in eluent. Cantitatea de adsorbant este calculata la circa 25 - 50 g pentru un gram substanta activa de separat. Dupa indepartarea solventului din coloana si spalare cu solventul ce va fi utilizat in separare, la partea superioara a coloanei se introduce proba sub forma unei solutii concentrate, dizolvata intr-un solvent bun. Se lasa sa se distribuie uniform pe suprafata coloanei, eventual prin adaus cantitati limitate de solvent. Dupa ce proba a patruns complet in coloana, se adauga un strat de aproximativ 1 cm de nisip, dupa care se incepe adaugarea de solvent (eluent) proaspat. Etapa corespunde developarii, cand componentele probei analizate se distribuie pe coloana in raport cu afinitatea fiecaruia fata de faza stationara. In urma elutiei cu solvent, compusii din amestec se pot separa sub forma de benzi colorate, de-a lungul coloanei, fiecare "banda" colorata putand fi sectionata si analizata ulterior, metoda fiind aplicabila amestecului de pigmenti de culori diferite (de ex. la separarea clorofilelor si pigmentilor insotitori, caroteni si xantofile). O alta metoda presupune elutia cu solventi pana cand eluantul aduce la capatul coloanei diverse fractii ce se colecteaza si se analizeaza sub aspectul compozitiei. Cea de a doua varianta se aplica la extractia b-carotenului din plante, a flavonelor din materiale vegetale, a izomerilor HCH, a fitosterolilor, peptidelor etc. Adsorbantul din coloana poate fi regenerat si reutilizat.
Pentru cresterea vitezei de elutie pe inaltimea coloanei, aceasta se poate atasa la un vas de trompa legat la vid sau se poate lucra sub aer comprimat (flash cromatography).
In prezent se folosesc coloane cu adsorbanti de mare specificitate, fata de care, compusii se separa prin inversie de faza.
1.2. Cromatografia plana
Cromatografia plana se caracterizeaza prin forma geometrica plana a suportului fazei stationare. Aceasta tehnica cromatografica include: cromatografia pe hartie (CH) si cromatografia pe strat subtire (CSS). Separarea se bazeaza pe distributia componentelor intre doua faze, una mobila lichida si una stationara. In cromatografia pe hartie (CH), faza stationara este fibra celulozica cu anumite caracteristici structurale si dimensionale. In cromatografia in strat subtire (CSS), faza stationara este silicagelul, alumina, poliamide ca nylon-6 si nylon-6,6, celuloza aminoetilata, amidonul etc., toate avand in masa un liant pentru formarea stratului subtire pe un suport inert (placute de sticla sau folii de aluminiu). Faza mobila este un amestec de solventi bine alesi.
Aceste metode prezinta avantaje ca: universalitatea, simplitatea, accesibilitatea si rapiditatea metodei, posibilitatea analizei simultane a mai multor probe, eficienta mare de separarea a substantelor, posibilitatea utilizarii pentru identificare a reactivilor corozivi sau a developantilor acizi.
Cromatografierea pe hartie sau strat subtire presupune developarea intr-un spatiu inchis - camera de developare (bac cromatografic) - cu atmosfera saturata de solventi. Developarea poate fi ascendenta sau descendenta, orizontala, circulara sau radiala. Camerele de developare utilizate pot fi cilindrice sau paralelipipedice. In practica se utilizeaza cuve rectangulare, de tip sandwich sau de alte forme, in care se introduce faza mobila (un solvent sau un amestec de solventi).
Modul de lucru
Hartia cromatografica are o structura poroasa speciala. Aceasta se decupeaza la dimensiunile cerute de bacul cromatografic si de numarul de spoturi care urmeaza a fi depuse. Daca n este numarul de spoturi, atunci latimea hartiei va fi (n+2) 2 cm, iar lungimea egala cu inaltimea bacului. Placile pentru strat subtire sunt pregatite in prealabil si activate. Ele au dimensiuni standard, iar adsorbantul se alege dupa natura componentilor si amestecul de solventi pentru elutie. Amestecul de solventi se introduce in camera sau bacul de cromatografie si se ataseaza capacul pentru a se ajunge la saturatie cu vapori.
Proba de analizat, dizolvata in general intr-un solvent, se aplica la baza placii sau hartiei cromatografice, la 10-20 mm de margine, cu ajutorul unei capilare calibrate sau cu o seringa speciala. Probele aplicate pe placi trebuie sa contina minim 0,01% concentratie in componentele de separat, insa nu trebuie sa fir prea concentrate, deoarece la concentratii mari separarea este difuza (spoturile nu sunt distincte) obtinandu-se numeroase suprapuneri sau asimetrii. Dupa aplicarea probelor, locurile de depunere aflate pe linia de start, trebuie uscate foarte bine, deoarece solventii de extractie remanenti pot influenta negativ separarea substantelor de analizat. Solventii volatili se pot indeparta cu usurinta, prin simpla agitare in aer a cromatogramelor, in timp de solventii cu puncte inalte de fierbere se indeparteaza prin incalzirea placilor cromatografice la temperaturi controlate, care sa nu afecteze structura placilor si componentii din proba. Dupa evaporarea solventului de extractie, cromatoplaca se introduce in camera de developare care contine faza mobila (fig.1.).
|
|

Fig. 1. Detalii privind aplicarea cromatografiei plane:
(a) bac cromatografic (1) cu: nacela pentru amestec de solventi (2), placa sau hartia suport (3), bagheta de sprijin(4), capac etans (5); (b) placa sau hartia cromatografica; (c) cromatograma developata si distante folosite in calculul Rf - ului.
Dupa ce developantul a atins distanta de deplasare prevazuta (marcata prin frontul developantului) placa sau hartia cromatografica se scoate, se usuca si se trece la detectia componentilor. Revelarea este operatia de punere in evidenta a zonelor separate, corespunzatoare diferitilor componenti separati. Procedeele de vizualizare pot fi: fizice - expunerea cromatogramelor la lumina UV; chimice - reactii de culoare efectuate prin stropirea placii cu reactivi chimici; fizico-chimice - stropirea cu reactivi chimici urmata de expunere la lumina UV; biochimice - bioautografia, reactii enzimatice. Spotul poate fi analizat cantitativ prin decupare sau razuire urmata de extractia componentului intr-un volum determinat dintr-un solvent potrivit sau direct prin densitometrie optica.
Modul general de evaluare a rezultatelor separarii este dat de valoarea Rf, care reprezinta raportul dintre distanta xi de la linia de start pana la centrul componentului separat si distanta H de la linia de start la frontul fazei mobile:
Rf = xi / H
Pentru conditii identice de cromatografiere pe hartie si strat subtire (conditii standard), Rf-ul devine o constanta de identificare a fiecarui component separabil.
Metoda se aplica curent in analiza aminoacizilor din hidrolizate proteice, a zaharurilor si glicozidelor din produse naturale, in chimia heterociclurilor, sterolilor, carotinoidelor si multe altele.
Cand separarea componentelor nu a fost totala sau convenabila, se apeleaza la cromatografia bidimensionala. Aceasta consta in reeluarea cromatogramei pe directie perpendiculara pe prima. Practic, in bacul cromatografic cu dimensiuni adecvate, se introduce cromatograma uscata si nedevelopata, in aceiasi solventi sau intr-un alt amestec.
1.3. Gaz-cromatografia (GC)
Cromatografia de gaze este procesul prin care componentii unei probe de substanta vaporizata se distribuie intre o faza mobila gazoasa (gazul purtator) si o faza stationara, care poate fi un solid cu proprietati adsorbante (CGS) sau un film lichid activ depus pe un suport inert (CGL). Cromatografia gaz-solid are loc printr-un mecanism de adsorbtie iar cromatografia gaz-lichid are loc printr-un mecanism de repartitie.
In fig. 2 se prezinta schema de principiu a unui cromatograf, in general, constituit din unitatea de introducere a probelor si eluentilor (gaz purtator, lichide sub presiune in HPLC, solutii tampon in cromatografie de schimb ionic etc.), coloana cromatografica in care se afla faza stationara si sistemul de detectie-inregistrare a semnalelor furnizate de detector. Chiar daca principial intre cromatografe nu apar diferente mari, tehnicile de separare insa, le deosebesc fundamental.
|
|

Fig. 2. Principiul constructiv si functional al cromatografelor
In gaz-cromatografie eluantul este un gaz inert (N2, Ar, He).
Functionarea gaz-cromatografelor In principiu, proba evaporata este preluata in curentul gazului purtator si distribuita in coloana. La iesirea din coloana, in eluent se regasesc treptat, componentii in ordinea crescatoare a retentiei lor pe faza stationara. Cand in eluant apare un component, detectorul sesizeaza modificarea unei proprietati fizice (conductibilitate termica, ionizarea flacarii, absorbtie in UV etc.) pentru care furnizeaza un semnal preluat de un amplificator, apoi de computer si afisat sub forma de cromatograma (o succesiune de picuri) similara celei prezentate idealizat in fig. 3.
|
|
Fig. 3. Exemplu de cromatograma pe care s-au separat opt componenti (x1 pana la x8). |
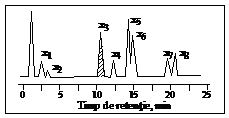
Timpii de retentie sunt caracteristici calitative pentru identificare prin comparare cu substante etalon, iar suprafata picurilor sunt caracteristici cantitative pentru componentii separati, ea fiind proportionala cu concentratia componentilor in amestec.
In tehnicile moderne, componentii separati intra intr-un spectrometru de masa (SM) atasat, care faciliteaza identificarea structurilor.
2. CARACTERIZAREA SPECTRALA A COMPUSILOR ORGANICI
Metodele spectrale sunt metode moderne foarte eficiente pentru determinarea structurii compusilor organici, precum si pentru determinarea compozitiei unor amestecuri de substante organice.
Iradierea unui atom sau molecule cu radiatii
electromagnetice din spectrul continuu, conduce, prin absorbtia de cuante
de energie cu anumite frecvente proprii, la trecerea de la starea
energetica fundamentala E intr-o stare excitata E',
diferentele energetice corespunzand relatiei: E' - E = DE = h n = h c/l = h c ![]()
Revenirea la nivelul energetic initial este
marcata prin emiterea unei cuante de energie egala cu cea
absorbita si poate fi inregistrata sub forma spectrelor de absorbtie, cu
ajutorul spectrofotometrelor. Spectrul de
absorbtie reprezinta redarea grafica a energiei absorbite
functie de frecventa (n), lungime de unda l, sau numar de unda (![]() = 1/l cm-1). Spectrele de absorbtie
sunt proprietati caracteristice ale substantelor.
= 1/l cm-1). Spectrele de absorbtie
sunt proprietati caracteristice ale substantelor.
Spectrele produse de atomii liberi sunt spectre de linii (spectre atomice), excitarea atomica constand in tranzitii electronice la nivele de energie superioare.
Excitatiile moleculare, care au loc in urma absorbtiei de energie, pot determina atat schimbari in energia electronica (tranzitii electronice in UV-VIS)cat si in energia vibratorie si rotatorie a moleculei (spectre de vibratie-rotatie in IR). Tranzitiile vibratorii si rotatorii moleculare depind de structura moleculara si necesita absorbtii in diferite regiuni spectrale. Spectrele optice ale moleculelor organice apar sub forma de benzi si sunt in general mult mai complicate decat spectrele atomice de linii.
Trebuie subliniat faptul ca o cantitate discreta de energie este necesara pentru fiecare tip de tranzitie (creste din IR la VIS si UV), deoarece toate fenomenele sunt cuantificate. Deci, caracteristic moleculelor, in afara de nivelurile de energie electronica (cu numerele cuantice electronice n=1,2,3) sunt si nivelurile de energie vibratorie (v=0,1,2) si cele de energie rotatorie (j=0,1,2).
Deoarece fiecare fel de excitatie moleculara necesita o cantitate specifica de energie, in spectrul electromagnetic absorbtiile apar in diferite regiuni, dupa cum se expune in urmatorul tabel:
Tabelul 2 Regiuni spectrale si tipuri de excitatie
|
Regiunea |
Lungimea de unda |
Energia excitatiei, kcal |
Tipul de excitatie |
||||
|
Raze gama, raze X |
<100 nm |
>>286 |
Penetrante; difractie de raze X |
||||
|
UV de vid |
100-200 nm |
|
Electronica |
||||
|
UV de cuart |
200-380 nm |
|
Electronica |
||||
|
Vizibil |
380-800 nm |
|
Electronica |
||||
|
IR, apropiat
|
0,8-2 m |
|
Alungire si deformare legaturi |
||||
|
IR, indepartat |
2-30 m |
|
Deformarea legaturii |
||||
|
Microunde |
1 cm |
|
Rotationala |
||||
|
Radiofrecventa |
metri |
|
Tranzitii de spin nuclear |
Pentru caracterizarea spectrala a substantelor organice se folosesc in mod curent spectrele de absorbtie in ultraviolet-vizibil (UV-VIS), infrarosu (IR) si rezonanta magnetica nucleara protonica (HRMN).
2.1. Spectrometria in UV-VIS
Toate substantele organice absorb radiatii in UV-VIS, respectand legea Lambert-Beer:
![]()
unde: El este extinctia la lungimea de unda a masuratorii sau densitatea optica (DO); Io si I sunt intensitatile radiatiei initiale si dupa traversarea solutiei de concentratie c prin stratul de grosime d (cm, drum optic); e este coeficientul molar de extinctie (cm2/mol sau L/mol cm), marime caracteristica pentru fiecare substanta si dependenta de l; daca c = 1 mol/L si d = 1 cm, e devine extinctie molara si arata cu cat se atenueaza intensitatea radiatiei incidente dupa traversarea substantei absorbante.
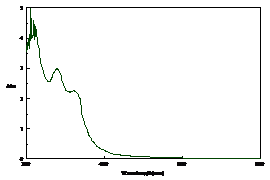
Absorbtia de energie se inregistreaza cu spectrometre in domeniul 190 - 1000 nm Spectrofotometrele pot fi utilizate fie pentru masuratori de extinctie fie pentru inregistrari de spectre UV-VIS. In fig. 4 este prezentat spectrul UV-VIS al acidului cinamic, activ in domeniul UV si fara absorbtie specifica in VIS, deoarece substanta este incolora.
|
|
Fig. 4. Exemplu de spectru UV-VIS |
Spectroscopia UV-VIS se aplica in doua directii distincte:
a) studiul structurii substantelor pe baza spectroscopiei empirice in UV;
b) determinarea concentratiei solutiilor substantelor care respecta legea Lambert-Beer (nu interactioneaza cu solventul, nu se asociaza etc.).
a) Spectroscopia empirica in UV
Metoda se bazeaza pe compararea spectrelor unor serii mari de compusi organici inruditi structural si pe observarea modificarilor produse de grupe functionale noi introduse in molecule sau la schimbarea solventului. S-a ajuns astfel sa se poata atribui diferitele benzi din spectre, unor grupe de atomi din molecule sau anumitor tipuri de tranzitii electronice.
Tranzitiile electronice pot avea loc dintr-un orbital de legatura s sau p ocupat cu electronii legaturii sau cu o pereche de electroni neparticipanti (n), in orbitali de antilegatura s* sau p*, liberi de electroni in stare fundamentala, dar care pot accepta electroni in stare excitata. In fig. 4 se prezinta tranzitiile electronice posibile in domeniul UV-VIS.
|
|
Fig. 5. Tranzitii electronice la absorbtia de energie in domeniul UV-VIS |
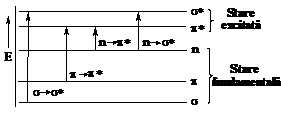
Energia cuantelor din UV-VIS variaza de la 120 la 600 kJ/mol, ceea ce determina tranzitii electronice, simultan cu modificari ale nivelelor vibratorii si rotatorii care largesc benzile de absorbtie. Cu cat electronii sunt mai mobili, cu atat energiile de excitare pentru tranzitie sunt mai mici si substanta va absorbi la valori l mai mari.
In hidrocarburi saturate sunt posibile doar tranzitii s s* care au loc sub 180 nm. Deci, alcanii si cicloalcanii nu sunt activi in UV.
Compusii saturati cu heteroatomi O, N, S, halogeni etc., pot prezenta in plus si tranzitii n s* sesizabile intre 200 si 250 nm.
Compusii nesaturati continand duble legaturi: >C=C< >C=O, >C=N N=N si N=O absorb la l mult mai mari de 200 nm datorita tranzitiilor p p*, in special, dar cu o importanta contributie si a celor de tip n p*. Aceste grupe functionale se numesc cromofori (purtatori de culoare).
Fenomenul de deplasare a absorbtiei spre lungimi de unda mai mari se numeste efect batocrom (inchiderea culorii sau deplasare spre rosu). Trebuie subliniat ca tranzitiile p p* in sisteme polienice conjugate deschise sunt mai intense decat in cele ciclice si aromatice. Fenomenul de conjugare a grupelor cromofore cu sisteme polienice si inele aromatice induce efecte batocrome extrem de importante.
Alteori, grupele functionale pot actiona prin deplasarea absorbtiei spre lungimi de unda mai mici (efect hipsocrom) sau prin modificarea intensitatii absorbtiei, prin cresterea (efect hipercrom) sau micsorarea acesteia (efect hipocrom)
Cu cat o molecula este mai complexa, cu atat si spectrul UV este mai complicat datorita multiplelor tranzitii electronice posibile la energii de excitare mult diferite.
Spectrele UV se inregistreaza in solventi inactivi in domeniul cercetat (hexan, ciclohexan sau alt alcan in UV de vid si respectiv, CCl4, metanol, apa etc., in UV de cuart), la concentratii de ordinul 10-2 10-4 M.
Aplicatii practice. 1. Se vor inregistra spectrele in UV-VIS, spre comparatie, la o substanta incolora (benzen, acetona etc.), alta colorata (nitrobenzen, -caroten etc.).
Se fac comentarii asupra diferentelor de absorbanta (e) si lmax din cele doua regiuni ale spectrului (UV si VIS), comparand o substanta colorata cu alta incolora, dar apropiata ca structura a catenei de C.
Aceleasi spectre se vor inregistra inainte si dupa purificarea unor probe.
Inregistrari de spectre se vor face si pentru substantele preparate in laborator.
b) Aplicatii ale spectroscopiei UV-VIS in dozarea substantelor
Substantele care respecta legea Lambert-Beer, prezinta dependenta liniara intre densitatea optica si concentratie: E = e c l. In felul acesta, se pot doza atat substante incolore, dar cu absorbanta specifica in UV, cat si substante colorate care absorb specific in vizibil. Cele mai numeroase aplicatii analitice se regasesc in domeniul substantelor colorate.
Masuratorile analitice in vizibil se fac la lungimea de unda a culorii complementare, a carei valoare exacta se stabileste experimental. Pentru aceasta, se parcurg urmatoarele etape:
- se stabileste in spectrul de absorbtie banda de absorbtie a substantei si influenta solventului sau a amestecului de solventi;
- se determina grafic lmax pentru care se obtine un emax pe curba e = f(l
- odata stabilit lmax, se verifica liniaritatea functiei E = f(c), care va arata si limitele de concentratie la care se lucreaza in solventul ales;
- in acest scop se prepara solutii cu concentratii descrescatoare de la 0,1 M la 10-3 M (se aleg cat mai multe puncte) si se citesc extinctiile fata de solvent si eventual adausul de reactivi care se gasesc si in proba cercetata, in felul acesta anulandu-se contributia lor la valoarea extinctiei inregistrate.
Culorile fundamentale si cele complementare se dau in tabelul 3.
Tabelul 3. Spectrul vizibil si lungimile de unda corespunzatoare
|
l, nm |
Culoare spectrala |
Complementara |
l, nm |
Culoare spectrala |
Complementara |
|
|
violet |
galben-verde |
|
galben-verde |
violet |
|
|
albastru |
galben |
|
galben |
albastru |
|
|
albastru-verde |
portocaliu |
|
portocaliu |
albastru-verde |
|
|
verde-albastru |
rosu |
|
rosu |
verde-albastru |
|
|
verde |
purpuriu |
|
|
|
Toate probele se efectueaza in cuve de aceeasi grosime (l), deci radiatia strabate acelasi drum optic. Graficul, E=f(c), va fi dreapta de etalonare, pe care aparatele moderne o traseaza automat.
Daca nu rezulta o dreapta, inseamna ca substanta nu respecta legea Lambert-Beer si deci, nu poate fi folosita pentru analiza chimica. Se va mai observa ca la concentratii prea mici si prea mari, apar abateri de la liniaritate datorita relatiei logaritmice dintre I si E. Aceste valori se exclud.
Dozarile in UV se fac dupa aceeasi metodologie.
2.2. Spectrometria in IR
Spectroscopia in infrarosu este o metoda deosebit de utila pentru stabilirea structurii compusilor organici, punand in evidenta atat natura legaturilor C-C, C-H, X-H (in care X = N, O, S, B . ) cat si natura gruparilor functionale heteroatomice. Identificarea grupelor functionale din compusii organici prin spectroscopia IR depinde de observatia experimentala ca gruparile functionale absorb la frecvente unice specifice fata de scheletul hidrocarbonat, la care sunt atasate.
Prin absorbtie de energie de vibratie din domeniul infrarosu, nucleele atomice dintr-o molecula intra in stare de vibratii normale. O vibratie normala moleculara este aceea in care toate nucleele vibreaza cu aceeasi frecventa, de obicei in faza intre ele. Daca intr-o molecula sunt n atomi exista intotdeauna (3n-6) vibratii normale pentru moleculele neliniare, si (3n-5) vibratii normale intr-o molecula liniara.
Caracteristic absorbtiei in IR este faptul ca pentru a se putea observa o tranzitie este necesar ca momentul dipolar al moleculei sa varieze in timpul vibratiei, deci sa apara un dipolmoment temporar.
Excitatiile in IR determina: vibratii de alungire a legaturilor (vibratii de valenta), , care pot fi simetrice si antisimetrice, si vibratii de deformare a unghiurilor de valenta, . Este mai usor sa se deformeze o molecula prin torsiune (variatia unghiului de valenta) decat prin alungirea legaturilor; de aceea energia si frecventa vibratiilor de deformare sunt intotdeauna mult mai joase ca ale vibratiilor de alungire a legaturilor.
Studiul asupra unui numar mare de molecule organice a condus la impartirea domeniului IR in urmatoarele regiuni:
regiune a excitatiilor de alungire (650 - 4000 cm-1);
regiunea excitatiilor de deformare (500 - 1550 cm-1);
regiunea excitatiilor rotationale (< 800 cm-1).
In general sunt active in IR vibratiile nesimetrice care creeaza un dipol temporar.
Benzile de absorbtie in IR au intensitati diferite si in general frecventa lor variaza in functie de natura legaturii, astfel:
a. legaturile simple continand H: C-H, N-H, O-H, S-H, B-H dau absorbtii de alungire in regiunea IR: 3700 - 2200 cm-1;
b. legaturile triple: CsC, CsN, aproximativ intre 2300 - 2000 cm-1;
c. dublele legaturi ca: C=O, C=N, C=C, N=O, dau absorbtii intre 1850-1480 cm-1.
Ca o regula generala, legaturile mai polare dau benzi de absorbtie mai puternice
Una din aplicatiile curente ale spectroscopiei IR este la identificarea substantelor (solide, lichide sau gazoase). Fiecare substanta poseda un spectru in IR caracteristic, deosebit de al oricarei alte substante, un fel de "amprenta digitala" ("finger print") a sa. In tabelul anexat se prezinta absorbtiile caracteristice in IR, importante pentru elucidarea structurilor organice.
Tabelul 4. Frecvente de grup caracteristice substantelor organice
|
Vibratii |
Tipul de legatura |
Substante organice |
|
legaturi cu H |
||
|
3600-3650 cm-1 |
-O-H |
Alcooli monomeri |
|
|
R-O-H |
Legatura de hidrogen |
|
|
-N-H |
Amine si amide |
|
|
-C-H |
Acetilenic (Csp-H) |
|
|
-C-H |
Alchenic si aromatic (Csp2-H) |
|
|
-C-H |
Saturat (Csp3-H) |
|
|
-COO-H |
O banda larga dantelata fara maxim unic |
|
|
-C-H din CHO |
Banda ascutita si distincta |
|
|
-S-H |
Gruparea tiol |
|
|
-N-H |
Saruri de amoniu |
|
domeniul triplei legaturi |
||
|
cca 2200 cm-1 |
-CsC- |
Banda slaba, absenta in alchinele simetrice |
|
|
-CsN |
Gruparea nitril |
|
|
-C=C=C- |
Alene: (R2)H2C=C=CH2 |
|
domeniul dublei legaturi |
||
|
A. Grupari carbonil, >C=O, (valori 15 cm-1) |
||
|
1820 cm-1 |
R-CO-X |
Halogenuri acide |
|
|
(R-CO)2O |
Anhidride acide |
|
|
R-COOAr |
Esteri aromatici ai acizilor saturati |
|
|
R-COOR" |
Esteri ai acizilor saturati |
|
|
C=O |
Aldehide si cetone saturate |
|
|
C=O |
Ciclohexanona |
|
|
C=O,C=C |
Cetone monoconjugate (ex.acetofenona) |
|
|
R-CO-NH2 |
Amide primare |
|
|
|
Amide secundare |
|
|
|
Amide tertiare |
|
|
|
Cetone diconjugate (benzofenona) |
|
B. Alte legaturi p |
||
|
1640 cm-1 |
C=C |
Alchene,banda slaba |
|
|
C=C-C=C |
Alchene conjugate |
|
|
S=C=S |
Sulfura de carbon |
|
1550 si 1350 |
-NO2 |
Gruparea nitro,banda intensa |
|
|
C-C |
Alcani |
In fig. 6 se prezinta un exemplu de spectru IR in care s-au indicat cateva frecvente de grup caracteristice substantelor organice.
|
|

Fig. 6. Exemplu de spectru IR al unei substante organice cu diverse functiuni
Aplicatie. Se inregistreaza spectrul IR al unei substante organice cunoscute in solutie de CCl4 si se identifica frecventele de grup conform tabelului 4 sau cataloagelor de spectre IR.
Spectroscopia IR analitica se aplica la substante purificate in prealabil pentru eliminarea oricaror interferente substanta de baza-impuritati.
2.3. Rezonanta magnetica nucleara (RMN)
Dintre toate metodele fizice, rezonanta magnetica nucleara (RMN) ofera cele mai complete informatii asupra structurii substantelor organice.
1) Bazele teoretice ale RMN
Metodele de rezonanta magnetica se bazeaza pe proprietatea nucleelor cu numar impar de protoni sau neutroni sau ambele, de a prezenta un spin nuclear diferit de zero. Astfel, proprietati magnetice apar la 1H, 13C, 15N, 17O, 31P (ale caror nuclee au spinul nuclear I = 1/2), pe cand nucleele la 12C, 16O, cu numar par de protoni si neutroni,cu spin opus, momentul magnetic de spin nuclear se compenseaza, devenind nul. De aceea prezenta 12C si 16O in compusi organici nu se poate identifica prin RMN.
Nucleele, particule incarcate electric aflate in miscare, se comporta ca niste magneti minusculi, care genereaza un camp magnetic, caracterizat prin momentul magnetic de spin - i. Momentul magnetic al nucleelor se poate pune in evidenta prin interactiunea sa cuantificata cu un camp magnetic exterior. Un nucleu cu numar cuantic de spin I, introdus intr-un camp magnetic extern (H0) poate adopta (2I+1) orientari. Astfel, 1H, 13C, 19F cu I = 1/2, pot adopta doua orientari: una paralela cu campul extern H0 si una antiparalela, prima fiind mai saraca in energie (fig. 7).
|
|
Fig. 7. Diferenta de energie intre orientarea paralela si antiparalela fata de campul magnetic exterior H0. |

In campul magnetic H0, nucleul se comporta ca un giroscop cu miscare de precesie in jurul axei sale (precesie Larmor). Frecventa miscarii de precesie n, este data de relatia:
n m H = g Ho /2p
unde: m este momentul magnetic nuclear si g - raportul giromagnetic.
Trecerea de la orientarea paralela la cea antiparalela se face cu absorbtia unei cantitati de energie, DE, dependenta de intensitatea campului exterior H0:
DE h n = h g Ho /2p de unde: n g Ho /2p
Deoarece momentul magnetic al nucleelor este foarte mic , diferentele de energie dintre cele doua orientari sunt si ele foarte mici (~10-3 Kcal.mol-1) si deci, si frecventele cuantelor absorbite vor fi de acelasi ordin de marime.
In cazul protonilor, in stare normala, exista un mic exces de protoni orientati paralel, deci mai saraci in energie. Trecerea 1H paraleli in 1H antiparaleli se face prin absorbtia unei cuante de radiatie electromagnetica a carei frecventa este egala cu frecventa miscarii de precesie n. Deci fenomenul de rezonanta are loc in momentul in care frecventa campului exterior perturbator este egala cu frecventa Larmor a substantei de cercetat. Cand se indeplineste conditia de rezonanta apare un semnal al carui intensitate este proportionala cu numarul de protoni care au absorbit cuante de frecventa n
In aparatele de RMN, conditia de rezonanta se realizeaza, fie variind, in limite inguste, intensitatea campului magnetic exterior Ho si mentinand constanta frecventa (baleiaj de camp), fie variind frecventa si mentinand campul Ho constant (baleiaj de frecventa). Majoritatea aparatelor sunt bazate pe primul principiu, mai usor de realizat tehnic.
2) Aparatura RMN si inregistrarea spectrelor
In fig. 7 se prezinta principiul constructiv al aparatului RMN. La inregistrarea spectrului RMN proba de analizat P, se aseaza intre polii unui magnet de camp Ho (in majoritatea aparatelor Ho = 14092 G, ce corespunde frecventei de 60 MHz). Radiatia electromagnetica cu frecventa constanta de 60, 100 sau 200 MHz se realizeaza cu un oscilator de radiofrecventa (G1), ce transmite frecventa n prin bobina ce inconjoara proba. Marind progresiv Ho se ajunge la conditia de rezonanta cand 1H din proba absorb energie, generand un semnal care este detectat (D), amplificat (A) si inregistrat (I).
|
|
Fig. 8. Schema de principiu a unui aparat HRMN: B - sursa de curent continuu sau redresor; G1 - generator de radiofrecventa; G2 - generator de curenti in "dinti de ferastrau"; P - eprubeta cu proba de analizat; NS - magnet ce realizeaza campul exterior Ho; D- detector;A - amplificator; I - inregistrator |
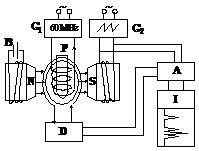
Inregistrarea semnalelor de rezonanta se face treptat, pe masura cresterii intensitatii campului exterior Ho. Astfel rezulta un spectru HRMN.
3) Interpretarea spectrelor 1HRMN.
Nucleele atomilor de hidrogen (protonii) ale unei substante plasata intr-un camp magnetic de intensitate H0, absorb radiatii electromagnetice de o anumita frecventa (cea de rezonanta), iar absorbtia de energie este inregistrata grafic sub forma unor semnale, rezultand spectrul RMN:
Spectrul de 1HRMN ne furnizeaza trei grupe de informatii despre:
a. deplasarea chimica D, care indica tipurile de protoni (alchilic, vinilic, acetilenic, aromatic, benzilic etc.);
b. numarul atomilor de hidrogen echivalenti, fiecare cu semnal propriu;
c. cuplajele spin-spin sau scindarea semnalelor.
a). Numarul atomilor de hidrogen care determina un semnal
Marimea caracteristica pentru intensitatea semnalului RMN este aria de sub curba care se numeste pic (fig. 9). Suprafata picului este proportionala cu numarul atomilor de hidrogen care il determina si se poate inregistra odata cu spectrul, sub forma curbei integrale. Numarul de semnale din spectru indica tipurile diferite de protoni din molecula. Toti protonii moleculei care au vecinatati identice din punct de vedere chimic si magnetic intra in rezonanta la aceeasi frecventa si vor da acelasi semnal. Acestia se numesc protoni echivalenti.
|
|
Fig. 9. Interpretarea unui semnal din spectrul HRMN (pic), corespunzator intensitatii semnalului protonilor echivalenti al carui numar se determina din curba integrala. |

De exemplu, in clorura de etil (CH3-CH2-Cl), notand atomii de H ai grupei metil cu a si pe cei ai grupei metilen cu b, apar doua semnale, ale caror intensitati corespund la 3Ha si respectiv, 2Hb. La clorura de n-propil (CH3-CH2-CH2-Cl) apar 3Ha, 2Hb si 2Hc (legati de -Cl, deci alte vecinatati), apar 3 semnale, cate unul pentru fiecare tip de protoni. La clorura de izopropil ((CH3)2CHCl) apar numai doua semnale, unul pentru cei 6 atomi de H din grupele metil si unul pentru protonul grupei metin. La alcoolul etilic (CH3-CH2-OH) apar tot 3 semnale ca si la 1-clorpropan, pentru ca sunt 3 tipuri de H neechivalenti, la metil, metilen si hidroxil.
b). Deplasarea chimica - tipuri de protoni
Daca la spectrele IR unitatea de masura curenta pentru determinarea pozitiei benzilor de absorbtie era micronul sau numarul de unda (cm-1), la RMN nu se poate stabili o valoare universal valabila, deoarece rezonanta este functie de intensitatea campului magnetic. Fara alte influente ar fi de asteptat ca toti protonii sa dea un singur semnal, la aceeasi frecventa. In realitate se observa ca diferitele categorii de protoni din compusii organici, intra in rezonanta pe rand, generand semnale la intensitati de camp diferite. Desi diferentele sunt extrem de mici (de cateva milionimi - parti pe milion - ppm - din valoarea absoluta a campului), ele sunt semnificative si constituie baza utilizarii spectroscopiei RMN pentru identificarea diferitelor categorii de protoni din moleculele compusilor organici. Originea acestor diferentieri se datoreaza existentei invelisului de electroni din jurul nucleului de hidrogen. Acesti electroni, sub influenta campului magnetic exterior, creeaza si ei un camp magnetic indus H', opus campului exterior (efect de ecranare diamagnetica). Acest efect de ecranare difera de la o categorie de protoni la alta, in functie de densitatea de electroni din jurul nucleului. Aceasta explica esalonarea semnalelor la diferite valori ale campului Ho.
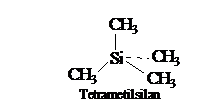
Practic, s-a constatat ca semnalul de rezonanta al unui nucleu ecranat apare la intensitati mai mari ale campului fata de un nucleu neecranat. Acest efect se numeste deplasare chimica pentru ca depinde de electroni, deci de caracterul chimic al atomilor. In practica, deplasarea chimica (D) se raporteaza la semnalul unei substante etalon sau standard (S), care se adauga in solutia probei de cercetat (standard intern). Acest standard este terametilsilanul (TMS), cu 12 atomi de H echivalenti, complet ecranati ce dau un singur semnal la Ho mai inalt decat protonii majoritatii substantelor organice. Ca substanta etalon se mai poate folosi si hexametildisiloxanul, care la temperatura inalta nu este volatil.
Deplasarea chimica reprezinta diferenta, intre pozitia semnalului probei si a standardului, exprimata fie prin intensitatea campurilor (D = H - HS [Hz]), fie prin frecventa de rezonanta (D n nS [Hz]). Diferenta D dintre protoni ajunge pana la 2000 Hz pentru frecventa uzuala a aparatului de 100 MHz.
Pentru a avea o marime proprie deplasarii chimice, care sa fie independenta de caracteristicile aparatului, deplasarea D se raporteaza la Ho sau la no, cand rezulta deplasarea chimica d, marime adimensionala, tabelata pentru numeroase substante organice:
![]()
Multiplicarea cu 106 se face pentru a obtine valori usor de exprimat.
Conform celor de mai de sus, o deplasare D = 436 Hz (la HCCl3), fata de TMS, masurata cu un spectrometru care lucreaza la 60 MHz corespunde la o deplasare chimica d = (436 Hz/60 106 Hz) 106 = 7,27 ppm. Daca nu s-ar fi inmultit cu 106 ar fi rezultat un numar aproape lipsit de semnificatie.
Conventional s-a atribuit semnalului TMS valoarea d = 0, iar protonilor din restul substantelor organice, d > 0. Intr-o alta scala, protonilor din TMS li s-a atribuit t = 10 si celorlalti protoni, t < 10 ppm. Prin urmare, t d = 10 si cele doua scari devin comparabile.
Se observa ca inmultind deplasarea chimica d in ppm cu frecventa de emisie a aparatului in MHz, se obtine D in Hz (D d (ppm) n (MHz).
In fig. 10 este prezentat spectrul RMN al tert-butanolului.
|
|

Fig. 10. Spectrul RMN (cu picuri) al tert-butanolului si curba integrala a semnalelor
Cand o molecula este plasata intr-un camp magnetic exterior se produce excitarea electronilor de legatura care, prin miscarea lor induc un camp magnetic circular suplimentar (indus) care poate : fie sa se opuna campului exterior, fie sa se insumeze cu acesta.
De exemplu, la acetilena campul magnetic circular indus, creat prin excitarea electronilor legaturii C-H este opus campului magnetic si deci semnalul apare la campuri mai intense; spunem ca electronii ecraneaza protonul (d = 2,3 - 3,9ppm.) .
In cazul benzenului, campul magnetic circular suplimentar are acelasi sens cu campul magnetic H0 , (se insumeaza) si deci rezonanta protonului (semnalul) are loc la un camp exterior mai slab, deci la deplasari mai mari (d = 6-8 ppm.)
Dezecranarea depinde si de efectul inductiv al altor grupe legate de atomul de carbon.
c) Cuplaje spin-spin si scindarea semnalelor
Spectrele RMN se complica deoarece campul magnetic real la care se supune un proton este modificat de campul creat de protonii vecini. Atomii invecinati pot adauga sau scadea campurile lor la campul exterior Ho. In consecinta protonul poate prezenta un semnal scindat in mai multe linii. Cu alte cuvinte, despicarea semnalului se datoreaza interactiunii dintre spinul nuclear al protonului si spinul nuclear al protonilor de la C vecin. Interactiunea numita cuplaj spin-spin se face prin intermediul electronilor de legatura datorita tendintei lor de a-si cupla spinul cu al celui mai apropiat proton. Aceasta interactiune se transmite prin electronii de legatura la protonul urmator.
Cuplajul spin-spin se manifesta prin scindarea picului astfel:
- un proton va scinda picul protonilor de la carbonul adiacent intr-un dublet;
- doi protoni vor scinda picul protonilor de la carbonul adiacent in triplet;
- trei protoni vor scinda picul protonilor de la carbonul adiacent in cuartet.
Deci n protoni vor scinda picul protonilor de la carbonul adiacent in (n+1) semnale (n = numarul de protoni vecini identici). Valoarea (n+1) se numeste multiplicitate.
De exemplu: CH3(b)-CH2(a)-Cl 2Ha-cuartet si 3Hb-triplet.
CH3(a)-CH(b)Cl-CH3(a): 6Ha-dublet si 1Hb-septuplet.
CH3(c)-CH2(b)-CH2(a)-Cl: 2Ha-triplet; 2Hb-sextet si 3Hc-tiplet.
CH3(c)-CH2(b)-OH(a) 1Ha-triplet; 2Hb-cvintet si 3Hc-triplet.
Interactiunea spin-spin si scindarile nu vor avea loc daca protonii Ha si Hb sunt inconjurati (ecranati) de densitati electronice asemanatoare
Constante de cuplaj. Distanta dintre maximele benzilor unui semnal RMN se numeste constanta de cuplaj (J). Ea este independenta de intensitatea campului H si se exprima in Hz. J este o masura a interactiunii sau a gradului de cuplare a spinilor protonilor care dau semnal cu protonii vecini. Cateva exemple de cuplaje spin-spin si semnificatia constantelor de cuplaj corespunzatoare apar in fig. 11.
|
|
Fig. 11. Exemple de multiplicare a semnalelor prin cuplare spin-spin si constantele de cuplaj aferente |

Pentru o imagine mai clara asupra cuplarii spinilor si intensitatile de interactie spin-spin, in fig. 12 se prezinta spectrul HRMN al etanolului.
|
|
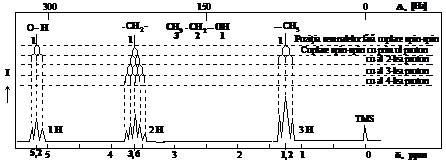
Fig. 12. Spectrul RMN de inalta rezolutie al etanolului (frecventa de emisie 60 MHz)
In literatura se dau valorile de referinta ale deplasarilor chimice si ale constantelor de cuplare J, care servesc la interpretarea spectrelor HRMN.
| Contact |- ia legatura cu noi -| | |
| Adauga document |- pune-ti documente online -| | |
| Termeni & conditii de utilizare |- politica de cookies si de confidentialitate -| | |
| Copyright © |- 2025 - Toate drepturile rezervate -| |
|
|
|||
|
|||
|
|||
Proiecte pe aceeasi tema | |||
|
| |||
|
|||
|
|
|||